Data collection
MiCoDa grew from an assessment (Jurburg et al. 2020) of the availability of 16S rRNA gene sequence data. We evaluated whether published microbiome studies had made their data available in public sequence repositories, particularly NCBI, EBI, and DDBJ databases.
First, we selected all publications in 17 microbiome/microbial ecology journals between (Annals of Microbiology, Applied and Environmental, Microbiology, BMC Genomics, BMC Microbiology, eLife, Environmental Microbiology, Environmental Microbiology Reports, FEMS Microbiology Ecology, Frontiers in Microbiology, ISME Journal, Journal of Applied Microbiology, Journal of Microbiology, Journal of Microbiological Methods, Journal of Microbiology Korea, mBio, BMC Microbiome, and Nature Microbiology), published between 2015 and 2019. In total, we selected 26,927 articles.
MiCoDa samples have all been sequenced in the hypervariable region between basepairs 515 and 806, to increase comparability.
Then, we designed custom parsing algorithms to identify accession numbers, the sequencer used, the 16S rRNA gene region sequenced, and specifically, whether the universal primers 515F-806R had been used. F6or the 515F primer, we captured any combination of the occurrence of 515F(wd) or F(wd) 515 which was separated by an arbitrary number of white spaces in addition to barcoded versions of the original and the modified 515F primer. For the 806R primer, we exhausted all variations of this primer analogously. Our procedure also captures non-minimal mentions (i.e, 806Rb). We identified 635 studies which used these primers, and evaluated the availability of their sequence data as described in Jurburg et al. 2020.
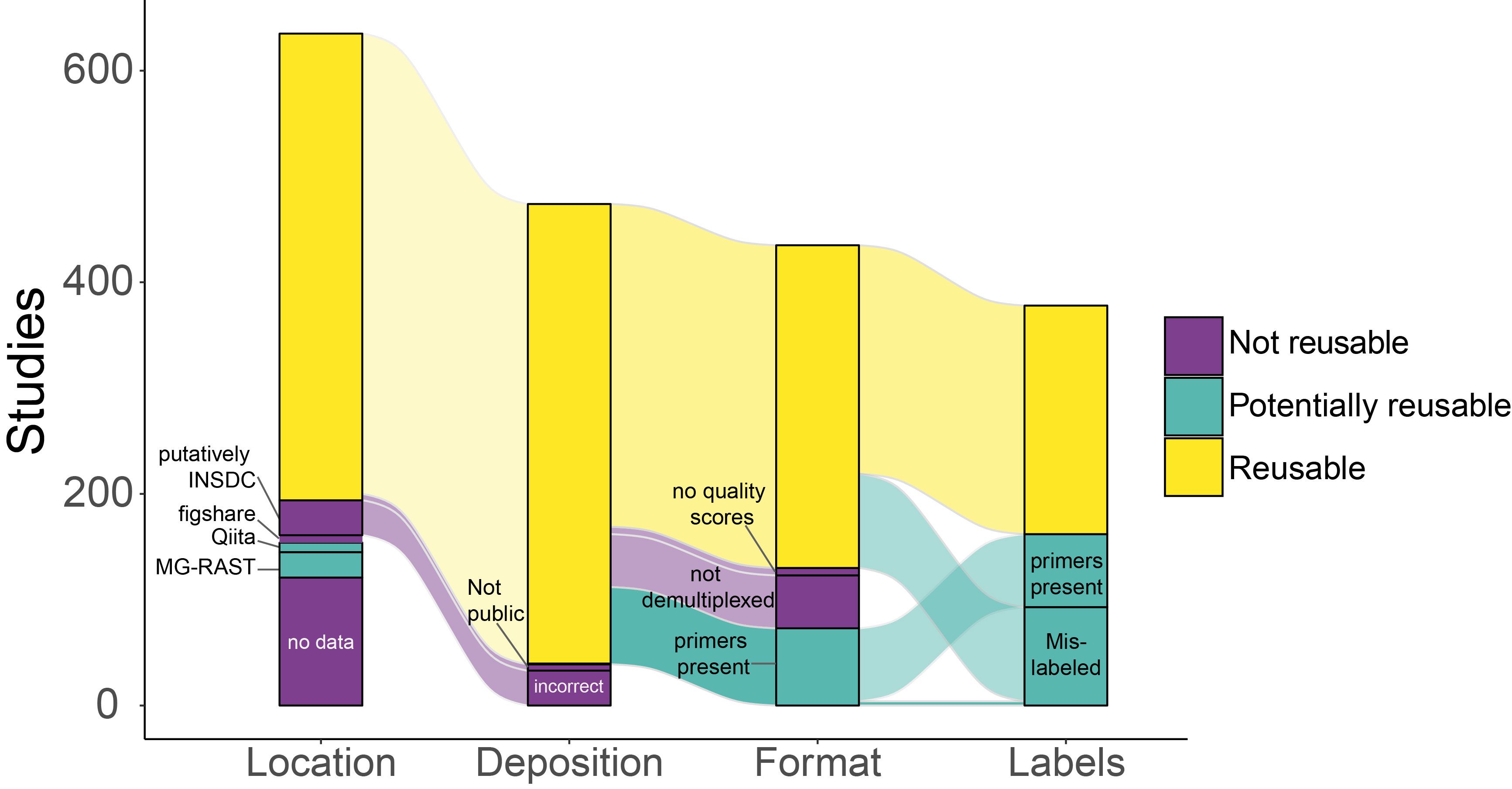
MiCoDa Version 1 includes data from 254 studies which had reusable data, did not contain primers, and were not mislabeled (i.e., the yellow category in the last column)
MiCoDa Version 1 includes 254 studies which had reusable sequence data (i.e., the data was retrievable from NCBI in a non-corrupted format) and did not have primers. In Version 1, we further manually excluded all samples which were not observational or experimental controls (i.e., blanks, mock communities, or treated mice). We also excluded samples which did not have sufficient metadata archived or in the publication’s materials to satisfy minimum MiCoDa metadata standards. We included metadata on the sample's environment by categorizing all environments into a four-level hierarchy (see Ontology). We also collected information on the laboratory techniques used (material and amount the DNA was extracted from, the DNA extraction protocol and the sequencer used), as well as the location of data collection (country, region, latitude and longitude). If the microbiome's environment was a host organism, we included information on taxonomy, the host's common name and Latin name, and whether the sample was obtained from the host's gut. Finally, to facilitate data reuse and the crediting of data providers, we collected publication information including the original article's DOI, journal, title, year of publication and as well as the abstract.
Additionally, we reprocessed all publicly available sequence data from the Earth Microbiome Project (EMP) and included it in the database as a collection. Publicly available EMP metadata was modified slightly to merge with the MiCoDa-wide ontology (see Ontology). As EMP data were already curated, they did not undergo the curation process of MiCoDa-specific samples and may include experimental treatments.
Sequence download and processing
Collected sequences were downloaded with NCBI's SRA tools, including prefetch, vdb-validate and fastq-dump tools. Downloaded sequences were processed using the dadasnake workflow, which uses the dada2 package to process amplicon sequences. As studies were checked for the presence of primers as part of the original assessment, they were not removed. To maximize comparability across samples from different studies which may have used single or paired-ended approaches, only forward reads for each sample were considered. First, each sample was rarefied (i.e., subsampled) to 5,000 reads. Sequences were trimmed to 90 base pairs in accordance with EMP recommendations (https://doi.org/10.1038/nature24621), and error models were trained on samples from each project separately. Taxonomies were assigned with the mothur classifier and the SILVA RefSSU_NR99 database v. 138.